LE CEFALEE AUTONOMICHE TRIGEMINALI
L. D’Alonzo, A. Negro, P. Martelletti
Centro Regionale di Riferimento per le Cefalee e Medicina Interna
Dipartimento di Scienze Cliniche e Molecolari
II Facoltà di Medicina e Chirurgia, Ospedale Sant’Andrea
Università “Sapienza”, Via di Grottarossa 1035, 00189 Roma
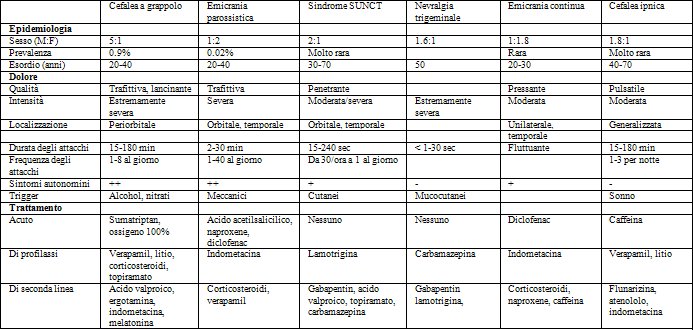
Le cefalee trigeminali autonomiche sono un gruppo di cefalee primarie caratterizzate da dolore episodico o cronico, unilaterale, localizzato alla testa e/o alla faccia, con elementi autonomici di accompagnamento [1,2]. L’“International Classification of Headache Disorders: 2nd edition (ICHD-II)” classifica le TACs come (1) cefalea a grappolo episodica o cronica (CH), (2) emicrania parossistica (PH) episodica o cronica (PH), e (3) attacchi nevralgiformi unilaterali di breve durata con iniezione congiuntivale e lacrimazione (SUNCT) [2]. L’emicrania continua (HC) è inclusa spesso in questo gruppo sebbene ICHD-II non colleghi queste entità nosologiche. Le TACs sono caratterizzate da attacchi di dolore di breve durata associati ad elementi lateralizzati che includono dolore, sintomi autonomini cranici, e dove presenti, sintomi emicranici, quali la fotofobia. Queste sindromi sono accomunate da diversi processi patofisiologici e condividono il medesimo iter diagnostico, pur differendo tra loro per la durata e la frequenza degli attacchi, e soprattutto per la risposta alla terapia, rendendo così fondamentale, al fine di una gestione ottimale, un’accurata diagnosi differenziale (TAB 1).
1. La cefalea a grappolo
1.1 Epidemiologia
CH e il tipo più comune di TACs, con una prevalenza nella popolazione generale di 0,5-1/1000 [3-12]. Il picco di esordio si registra in età giovanile (28-30 anni) ed è rara la sua comparsa in età infantile [13], sebbene vi siano stati riscontri di CH in bambini di 3 anni di età [14]. Esiste una predominanza per il sesso maschile [15], che negli ultimi anni è stata stimata in diminuzione probabilmente per un aumento di diagnosi corrette nel sesso femminile [16].
1.2 Presentazione clinica
L’ICHD-II suddivide CH in una forma episodica e una cronica [2]. Nell'80% dei casi, la malattia è episodica e può presentarsi con uno o due periodi di crisi all'anno, o può anche andare in remissione per molti anni prima di sviluppare un altro periodo di cluster. Nella forma cronica, che interessa circa il 20% dei pazienti con CH, gli attacchi sono continui per uno o più anni, con remissioni che non superano un mese [17]. CH è caratterizzata da attacchi ricorrenti (da 1 a 8 al giorno) di dolore unilaterale, straziante, di breve durata (dai 15 ai 180 minuti) accompagnato da segni di disfunzione autonomica, rappresentata da sintomi ipsilaterali quali iniezione congiuntivale e lacrimazione, congestione nasale o rinorrea, sudorazione della fronte e della faccia, edema palpebrale, arrossamento oculare, miosi e ptosi [2]. Questo pattern è molto importante per riconoscere la CH e differenziarla dagli attacchi emicranici. Nel 3% dei pazienti questi segni possono essere lievi o assenti [18], e in questi casi CH può essere diagnosticata se presente un senso di irrequietezza o agitazione durante gli attacchi. I sintomi spesso diminuiscono dopo i settant'anni di età. Tra i pazienti con CH è stata documentata frequentemente una forte abitudine al fumo [19]. CH mostra una ciclicità annuale e circadiana, con picchi di frequenza nei periodi di solstizio, in relazione ai cambiamenti nella durata della luce del giorno [20]. Gli attacchi possono presentarsi in un’ora precisa durante il ciclo di sonno, con una drammatica regolarità riferita dai pazienti. Il dolore è localizzato nel territorio della prima branca del nervo trigemino, su un lato della testa, e quasi sempre sullo stesso lato (raramente gli attacchi cambiano sede); può concentrarsi all'occhio o essere sovraorbitario e temporale, alcune volte esteso alla mascella, ai denti, all'orecchio o alla regione occipito-cervicale. La CH è una delle condizioni maggiormente dolorose conosciute dall'uomo, al punto che durante gli attacchi può essere presente un'ideazione suicidaria. Il dolore aumenta improvvisamente in pochi minuti e cessa altrettanto rapidamente. Nelle forme croniche severe o in quelle episodiche con numerosi attacchi giornalieri, può persistere un senso di disagio continuo sul lato interessato [21], anche se in genere i pazienti con CH sono completamente asintomatici tra gli attacchi. Durante gli attacchi, i pazienti tendono ad essere agitati, possono ondeggiare da un lato all'altro, colpirsi la testa, colpire oggetti con il pugno, o anche picchiare la loro testa contro il muro [22]. Questo comportamento è così tipico di CH che è accettato come un criterio ufficiale dalla ICHD-II [2]. L’irrequietezza può aiutare a differenziare CH dall'emicrania, nella quale i pazienti sono calmi ed evitano qualsiasi movimento. Gli attacchi dolorosi possono essere scatenati dall’alcool, dagli odori forti (soprattutto solventi e fumo di sigaretta) e dal sonno. Ciò che è sorprendente è che questi trigger sono efficaci solo durante le crisi, e quindi, nei periodi intercorrenti, i pazienti possono fumare e bere alcool senza che questo inneschi nuovi attacchi. Sebbene l’aura non sia frequente nella CH [23], più del 14% dei pazienti riportano sintomi ad essa correlati, che precedono il dolore facciale, quali disturbi visivi, motori o sensoriali transitori [17]. In circa il 50% dei pazienti sono presenti nausea, vomito, fotofobia e fonofobia, ma ciò non deve precludere la diagnosi di CH laddove gli altri criteri non siano soddisfatti. Sono stati descritti, anche se raramente, casi di “CH sine cefalea” con episodi di disfunzione autonomica facciale unilaterale senza dolore [24,25].
1.3 Diagnosi differenziale
La PH è più frequente nelle donne (80-90%) e si differenzia dalla CH per la minor durata e la maggior frequenza degli attacchi [26-28] e per una risposta positiva a una dose adeguata di indometacina [17]. Alcuni autori hanno suggerito un continuum tra PH e CH, data la loro sovrapposizione clinica e per il fatto che alcuni pazienti con CH rispondono bene all'indometacina. SUNCT è una sindrome molto rara e gli attacchi sono molto brevi e possono presentarsi fino a 300 volte al giorno. L’emicrania continua può essere confusa con la CH cronica, e in caso di dubbio, si dovrebbe ricorrere al test con indometacina, essendo la prima fortemente responsiva a questo farmaco [29,30] (TAB.1).
1.4 Terapia
I trattamenti acuti hanno lo scopo di alleviare il dolore durante gli attacchi, mentre quelli profilattici hanno lo scopo di ridurre il numero giornaliero degli attacchi.
1.4.1 Trattamenti acuti
La somministrazione per via orale agisce troppo lentamente ed è usata di rado. Per tale motivo sono preferiti trattamenti per via inalatoria, sottocutanea e intranasale. I due trattamenti acuti di maggiore efficacia sono costituiti dal sumatriptan sottocutaneo e dall'inalazione di ossigeno ad elevato flusso. Sumatriptan sottocutaneo 6 mg riduce efficacemente il dolore nella CH [31]. Dopo l'iniezione, il paziente può avvertire una vampata di calore e una costrizione toracica che scompaiono in alcuni minuti e sono seguiti da un rapido sollievo dal dolore. Sumatriptan [32,33] e zolmitriptan [34] intranasali sono impiegati nei pazienti che rifiutano le iniezioni. Le controindicazioni assolute al sumatriptan sottocutaneo sono la gravidanza, l'allattamento, le coronaropatie, gli ictus, e le arteriopatie periferiche. Le controindicazioni relative sono l'età (< ai 18 o > ai 65aa.), il fenomeno di Raynaud, l'allergia ai sulfamidici, il trattamento con un inibitore della ricaptazione della serotonina (SSRI). Il dosaggio massimo consentito in 24 h (Sumatriptan 12 mg sottocutaneo o 40 mg intranasale, zolmitriptan 10 mg intranasale) deve essere spiegato al paziente per evitare effetti collaterali pericolosi, come spasmi coronarici o arteriosi periferici. Sorprendentemente i pazienti con CH non sviluppano cefalea da abuso di farmaci (MOH) con i triptani così spesso come fanno gli emicranici [35]. Dato che MOH è più frequente nei pazienti con CH con una storia di emicrania, questi soggetti devono essere accuratamente monitorati per MOH, in particolar modo se CH è cronica e refrattaria [36]. L'inalazione di ossigeno ad alto flusso al 100% (da 10 a 15 l al minuto) è efficace e utile specialmente quando esistono controindicazioni all’uso dei triptani [37,38]. L’ergotamina e la diidroergotamina sono state usate nella CH [39]. Non devono essere somministrate con i triptani nello stesso periodo di 24 h, e condividono con questi le stesse controindicazioni, senza avere un'efficacia aggiuntiva. Per questi motivi sono utilizzate raramente. Nei casi di CH refrattaria, o in caso di controindicazione ai triptani, può essere provata l'applicazione intranasale di lidocaina, sebbene la sua efficacia non è bene accertata.
1.4.2 Trattamenti profilattici
L'uso continuo di trattamenti profilattici nella CH episodica non è raccomandato, dato che non esistono prove che ciò prevenga la crisi successiva. Quindi, la dose dovrebbe essere progressivamente ridotta dopo due o tre mesi fino alla sospensione, per poi essere ripresa in caso di ricomparsa degli attacchi. Il verapamil rimane il principale trattamento della CH episodica e cronica [40,41]. Il dosaagio varia tra 360 e 960 mg/die, e si dovrebbe raggiungere la massima dose tollerata prima di giudicarne l'efficacia. Gli effetti collaterali sono debolezza, fatica, edema agli arti inferiori, e blocco di conduzione cardiaca. Un ECG dovrebbe essere eseguito prima di iniziare la terapia e prima di ciascun incremento della dose quando questa supera i 480 mg/die [42]. Il topiramato 100 mg/die (range 25-200 mg) si è mostrato efficace nella CH [43,44]. Gli effetti collaterali sono torpore e formicolio alle estremità, perdita di peso, e difficoltà neurocognitive, tutti reversibili dopo la cessazione della terapia. Il litio 600-1200 mg/die è usato nella CH cronica [40,45,46]. Gli effetti collaterali includono tremori, insonnia, fatica, nausea e visione sfocata. L'intossicazione può essere severa e va sospettata velocemente se compaiono nausea, confusione, rigidità e difficoltà a camminare. A causa di questi effetti collaterali, il litio è impiegato sporadicamente, e solo in casi selezionati. Un breve ciclo con dosi decrescenti di prednisone può essere usato per trattare una crisi refrattaria al verapamil. Gli effetti collaterali a lungo termine della terapia cortisonica ne restringono l'impiego alla forma cronica di CH. Alcuni pazienti rispondono molto bene a methysergide 6-12 mg/die, che agisce sui recettori serotoninergici. La maggior limitazione è che triptani ed ergots non possono essere usati simultaneamente per il rischio di sindrome serotoninergica. Il principale effetto collaterale è la nausea, a volte severa. Methysergide può essere considerato se l'ossigeno da solo è usato come trattamento acuto. La maggior complicazione è la fibrosi retroperitoneale, polmonare o cardiaca dopo uso prolungato. Acido valproico [47], pizotifen, gabapentin [48,49], baclofen [50], e melatonina [51,52] hanno mostrato alcuni effetti nella profilassi e possono essere usati in pazienti selezionati come terapie di seconda linea. Gli approcci chirurgici hanno mostrato risultati variabili e complicazioni irreversibili come l'anestesia dolorosa [53-58].
L’“Hypothalamic deep-brain stimulation” (hDBS) è stata efficacia in alcuni pazienti refrattari [59-62], ma può essere completamente inefficace [63], e ha possibili complicazioni serie. Recentemente si è testata la neurostimolazione del nervo occipitale maggiore [42,64-66]. Essa è meno invasiva di hDBS ma più costosa, e non se ne conoscono gli effetti a lungo termine.
*TAB. 1 – Diagnosi differenziale delle cefalee autonomi che trigeminali
*
2. L’emicrania parossistica
La PH è caratterizzata da attacchi dolorosi severi e unilaterali localizzati alla regione orbitaria, sovraorbitaria, e temporale, accompagnati da uno o più elementi autonomici ipsilaterali. Gli attacchi nella PH sono più frequenti e di durata più breve (tra i 2 e i 30 minuti) rispetto alla CH ma a parte questo PH e CH hanno una presentazione simile. La caratteristica peculiare della PH è la cessazione completa della cefalea con l'indometacina e ciò la distingue dalle altre TACs [1,2].
2.1 Epidemiologia
PH rappresenta una piccola proporzione delle cefalee (1-3%) [67], e con una prevalenza nella popolazione generale di un caso per 50.000 persone. La forma cronica ha una prevalenza maggiore rispetto alla forma episodica (80 vs. 20%) [68]. La forma episodica è infatti rara, con meno di 30 casi riportati in letteratura [1,69,70]. A differenza di CH, PH ha una maggiore frequenza nel sesso femminile (F:M 1.6-2.36:1) [67,71]. Sebbene l’esordio sia più comune tra i 20 e i 30 anni [67,72], con un'età media di 34 anni [73], la PH può presentarsi a qualsiasi età, con un range compreso tra i 6 e gli 81 anni [73]; inoltre sono stati descritti casi di PH in bambini di tre anni di età [74-76]. Al momento non è stata dimostrata alcuna predisposizione genetica ed esiste solo un caso di PH che coinvolgeva una madre e la figlia [77]. In uno studio condotto su 74 pazienti con PH, l'80% non mostrava una storia familiare di nessun tipo di cefalea, il 15% aveva una storia familiare di emicrania, e il 5% aveva una storia familiare di altri tipi di cefalee e dolore facciale, incluse CH e la neuralgia trigeminale [71].
2.2 Presentazione clinica
Gli attacchi di cefalea nella PH sono tipicamente unilaterali. Solo in una minoranza di casi è presente un cambio di lato [78,79] ed è stato descritto un unico caso di attacchi di PH bilaterali [80]. Il dolore ha sede e maggiore intensità nella regione intra- e peri-orbitaria, temporale, mascellare, e frontale [2,68], con possibile coinvolgimento di collo, nuca e strutture orofacciali [68,72]. La maggioranza dei pazienti soffre della forma cronica, che è sostanzialmente incessante. Il 20% dei pazienti presenta la forma episodica che comporta attacchi di durata compresa tra le 2 settimane e i 4.5 mesi, con periodi di remissione di durata compresa tra 1 e 36 mesi [67,81]. La forma episodica in genere evolve nella forma cronica [82], sebbene sia stata descritta anche un’evoluzione in HC [83]. Meno frequentemente la PH cronica può regredire nella forma episodica [82]. La differenza tra le due forme è che la forma episodica di PH si presenta in periodi di durata compresa tra sette giorni e un anno ed è separata da un intervallo libero da dolore di almeno un mese. Al contrario, gli attacchi nella forma cronica si presentano per più di un anno senza periodi di remissione o con periodi di remissione di durata inferiore a un mese [2]. Gli attacchi di PH hanno in genere esordio e cessazione improvvisi, con una durata compresa tra i 2 e i 30 min, e potendo persistere per più di 2 ore [2,67,68]. La frequenza degli attacchi varia tra uno a 40 al giorno [67,71,84], presentandosi regolarmente a qualunque ora del giorno. Sebbene sia stata osservata un’associazione tra la fase di sonno REM e l’insorgenza di attacchi notturni, non vi è una loro predominanza come avviene nella CH [85]. Gli elementi autonomici cranici e facciali che più frequentemente si accompagnano agli attacchi di PH sono la lacrimazione, l’iniezione congiuntivale, la congestione nasale, e la rinorrea [2,67,68]. Meno frequentemente possono verificarsi edema palpebrale, ptosi, miosi e sudorazione facciale [2,68]. Sono stati anche descritti casi di PH accompagnati da elementi autonomici bilaterali [67], casi di PH senza elementi autonomici [86], ed elementi autonomici durante attacchi di PH senza presenza di dolore [79]. Durante un attacco di PH è comune anche la presenza di sintomi emicrania-simili, quali fotofobia, nausea, ed occasionalmente vomito e fonofobia [67,68,71]. Sebbene la maggior parte degli attacchi di PH siano spontanei, è stata riportata l’esistenza di trigger, quali l'assunzione di alcolici [67] e la rotazione meccanica o il piegamento della testa con una pressione esterna contro alcuni punti [87]. Inoltre, un attacco di PH può essere precipitato dall'applicazione di una pressione esterna ai processi trasversi delle vertebre C4 e C5, alla radice di C2, o al nervo occipitale maggiore [87,88].
2.3 Diagnosi
Diverse condizioni possono mimare la PH e una possibile difficoltà con la diagnosi differenziale di PH è la considerevole sovrapponibilità con CH. Queste possono essere differenziate dato che PH predilige il sesso femminile e presenta attacchi di cefalea più frequenti e di minor durata [2,89]. L'unico criterio basato sulla classificazione ICHD-II per la PH, rispetto alle altre TACs, è la responsività degli attacchi all'indometacina assunta per os. [2]. Un altro test diagnostico disponibile, seppure in attesa di una definitiva validazione, è l’“Indotest,” che prevede iniezioni i.m. di indometacina (50-100 mg), che comporta un’attenuazione sostenuta del dolore (in 493-668 min) [89,90]. Nei pazienti che non rispondono all'indometacina, l'iter diagnostico di PH dovrebbe essere riconsiderato [89], pur ricordando che sono stati riportati casi di PH non responsivi all'indometacina [71,91] (TAB 1)
2.4 Terapia
La profilassi degli attacchi di PH prevede l’uso di indometacina con una dose raccomandata di 75 mg/die per tre giorni, seguita, se necessario, da 150 mg/die per altri sei giorni [92]. Il dosaggio terapeutico è 50-125 mg/die, sebbene per trattare il dolore durante un’esacerbazione possano essere necessari 200-250 mg/die [93]. I pazienti non responsivi alle dosi maggiori difficilmente beneficiano di questa terapia. Nei pazienti responsivi, il dolore di solito scompare entro 24 ore con un range che oscilla tra poche ore e cinque giorni [67]. Quando l'assunzione di indometacina è discontinua, vi è spesso ricomparsa dei sintomi in un periodo compreso tra 12 ore e diversi giorni [94]. L’uso continuo di indometacina per un periodo di tempo indeterminato è la strategia terapeutica più indicata [95]. È stata però documentata una riduzione dell’efficacia terapeutica nel tempo [67,94] per ragioni non correlate alla tolleranza farmacologica [96]. Per prevenire i comuni effetti collaterali (dispepsia, anoressia, nausea, dolori addominali), e in genere nei trattamenti di lunga durata, possono essere prescritti analoghi delle prostaglandine (misoprostolo), antiacidi, inibitori dei ricettori H2 per l’istamina, o inibitori di pompa protonica [1,97]. Le controindicazioni all’uso di indometacina includono l’asma, l’anemia, l’insufficienza epatica e renale; in questi casi, così come nei non responsivi possono essere provati altri FANS (aspirina, diclofenac, e ketoprofene) [67,98,99] o l’inibitore COX-2, celecoxib [69,100]. I bloccanti dei canali del calcio come il verapamil hanno mostrato efficacia analoga a quella nella CH [99,101,102]. Altri farmaci come l’acetazolamide [91], il sumatriptan [103-105], e il topiramato [106], sono stati utilizzati con risulati variabili. Anche i corticosteroidi, dati i loro effetti antinfiammatori, sono stati impiegati nel trattamento di PH [98].
3. La sindrome SUNCT
SUNCT è un acronimo che sta ad indicare “attacchi nevralgiformi unilaterali di breve durata con iniezione congiuntivale e lacrimazione”. La sindrome è caratterizzata da dolore severo, strettamente unilaterale e di breve durata (tra 5 e 240 sec), localizzato alla regione orbitaria, sovraorbitaria, e temporale, accompagnato da elementi autonomici cranici e facciali ipsilaterali, tipicamente iniezione congiuntivale e lacrimazione. Gli attacchi sono di durata più breve e più frequenti rispetto alle altre TACs, con almeno tre attacchi al giorno fino a 200 attacchi al giorno, con ciascun attacco di durata compresa tra cinque e 240 secondi [2].
3.1 Epidemiologia
La sindrome SUNCT è estremamente rara, con circa 70 casi riportati in letteratura. A causa della sua rarità, la prevalenza e l'incidenza non sono note, sebbene sia probabilmente più comune di quanto suggerisca la letteratura, date la somiglianza e quindi la possibilità di errate diagnosi con la nevralgia trigeminale, la cefalea trafittiva, CH, e PH [107]. Diversi studi hanno riportato una prevalenza variabile in favore del sesso maschile (M:F da 1.3:1 a 3.75:1) [108, 109]. Sebbene l’esordio sia più comune tra i 35 e i 65 anni, con un'età media di 50 anni [109], la sindrome può presentarsi a qualunque età [68], con casi riportati di esordio precoce a cinque anni [110] o tardivo a 88 anni [111]. In letteratura è stato riportato solo un caso di SUNCT familiare, è resta ancora da chiarire se la sindrome possa avere un link familiare [112].
3.2 Presentazione clinica
Gli attacchi di cefalea nella sindrome SUNCT sono tipicamente unilaterali, senza cambio di lato nella maggior parte dei pazienti (88%), e localizzati più frequentemente sul lato destro (60%) rispetto al lato sinistro (36%) [108,109]. Sono stati anche descritti casi con cambi di lato tra gli attacchi [113,114], così come attacchi di SUNCT bilaterali [108,115]. Il dolore ha sede e maggiore intensità nella regione periorbitaria, temporale, e frontale, e occasionalmente coinvolge il collo, l'occipite, l'orecchio, il naso, la guancia, il palato e la gola [2,108,109,113,116,117]. Il 33% e il 21% dei pazienti con SUNCT riportano, inoltre, dolore localizzato, rispettivamente, alla branca mascellare del nervo trigemino e ai denti [118]. La qualità del dolore nella SUNCT è caratteristicamente di tipo nevralgico, e la sua intensità può essere moderata (15%), ma più frequentemente è intensa (65%) o straziante (20%) [108,109]. La maggioranza dei pazienti presenta periodi sintomatici che hanno durata di diversi giorni o mesi, con uno o due attacchi all'anno alternati da periodi di remissione. In genere il periodo di remissione ha durata di alcuni mesi, sebbene siano stati riportati casi di durata inferiore a una settimana o maggiore agli 8.5 anni [119]. I periodi di remissione, inoltre, tendono a ridursi con il tempo [108]. Sono stati descritti diversi casi che documentano l'esistenza di una forma cronica di SUNCT, con periodi sintomatici di durata tra gli 1 e i 17 anni [108]. Gli attacchi possono avere un pattern cronico fin dal loro esordio o evolvere da una forma inizialmente episodica a una forma cronica, o anche presentare un'alternanza tra le due forme [109]. E’ utile ricordare che l’ICHD-II non ha incorporato nei criteri di SUNCT una suddivisione tra le due forme, come invece è stato fatto per le altre TACs [2]. Gli attacchi di SUNCT hanno in genere un inizio improvviso e raggiungono l’intensità massima in 2 o 3 sec, con una durata media degli attacchi di 49 sec, con un range compreso tra 5 e 250 sec [108,113,120-122]. Sebbene rari, sono stati descritti anche attacchi di durata prolungata superiore alle 2 h. Gli attachi nella SUNCT hanno un esordio improvviso e una rapida cessazione, in genere con periodi liberi da dolore compresi dagli attacchi [108,109], sebbene siano stati descritti casi con disconfort interictale [122]. La frequenza degli attacchi varia da meno di uno al giorno a più di 60 all’ora [121], con ampie variazioni individuali e interindividuali [109]. Gli attacchi si presentano prevalentemente durante il giorno, con una tipica distribuzione bimodale nella mattina e nel pomeriggio o sera. Solo l’1.2% degli attacchi di SUNCT è notturno e di solito compare durante i periodi severi [123]. La quasi totalità degli attacchi esibisce elementi autonomici cranici e facciali ipsilaterali, quali iniezione congiuntivale e lacrimazione, con una prevalenza rispettivamente del 100% e del 94%, e meno frequentemente rinorrea (54%), congestione nasale (48%), edema palpebrale (26%), ptosi (12%), miosi (4%), sudorazione facciale (8%), e rossore (2%) [2,68,109]. Sono inoltre possibili elementi autonomici sistemici, come aumento della pressione arteriosa sistemica, riduzione della frequenza cardiaca, e iperventilazione [124,125]. In genere gli attacchi di SUNCT non si associano ad elementi emicrania-simili, quali nausea, vomito, fotofobia, fonofobia, e osmofobia [109,118]. Ciò nonostante, è stato riportato che il 49% dei pazienti con SUNCT presentano una storia di emicrania o hanno un parente di primo grado con emicrania [118]. La maggioranza degli attacchi iniziano spontaneamente sebbene possano essere innescati perlopiù immediatamente dalla pressione su alcune aree trigger comprese nella distribuzione del nervo trigeminale, e più raramente nell'area extra-trigeminale [107,108]. Un attacco può essere inoltre precipitato dal toccare o lavare la faccia o lo scalpo, dalla rasatura, dal mangiare, dallo spazzolarsi i denti, dal parlare e dal tossire [108]. In alcuni pazienti, la rotazione continua del collo è stata capace di ridurre o far cessare gli attacchi [108,126].
3.3 Diagnosi
L’ICHD-II ha recentemente classificato SUNCT, ma nonostante diversi report abbiano suggerito l’esistenza di forme episodiche e croniche, la classificazione non prevede questa distinzione [2]. Sono stati riportati casi di SUNCT preceduta da nevralgia trigeminale ad interessamento della branca oftalmica, e ciò potrebbe suggerire che la sindrome rappresenti in realtà una variante della neuralgia trigeminale [127-129]. Una variante di SUNCT è la sindrome SUNA, acronimo che indica “attacchi nevralgiformi unilaterali di breve durata con elementi autonomici cranici”. A differenza di SUNCT, SUNA può essere diagnosticata con solo uno degli elementi autonomici craniali o facciali, indipendentemente dalla presenza di rinorrea o lacrimazione. Inoltre, il dolore in SUNA può avere durata maggiore (< 10 min) rispetto a SUNCT [2]. La diagnosi di SUNCT poggia sulla valutazione dell'esordio rapido, la localizzazione, l'intensità, la qualità, e la durata del dolore, il pattern temporale degli episodi, i fattori scatenanti, e gli elementi autonomici associati [2]. Diverse condizioni rientrano nella diagnosi differenziale, quali la nevralgia trigeminali, la cefalea trafittiva, la cefalea ipnica, CH, e PH (TAB 1).
3.4 Terapia
La sindrome SUNCT è considerata relativamente refrattaria al trattamento [130]. Diversi farmaci impiegati per altri tipi di cefalea o sindromi dolorose si sono dimostrati poco efficaci o del tutto inefficaci; questi includono FANS e COX-2 inibitori (indometacina e nimesulide), analgesici (acetaminofene e oppioidi), agonisti della 5-idrossitriptamina (triptani, ergotamina, e diidroergotamina), betabloccanti (propranololo e timololo), agonisti alfa-adrenergici (clonidina), antistaminici, antidepressivi triciclici (amitriptilina e nortriptilina), calcio antagonisti (nifedipina, flunarizina, e diltiazem), litio, fenitoina, acido valproico e clonazepam [113,115,120,126,127,130-135]. Alcuni farmaci, specialmente i calcio antagonisti verapamil e amlodipina, hanno addirittura peggiorato i sintomi [1,108,119]. Anche l'inalazione di ossigeno al 100% non ha dato risultati positivi [50]. Al momento l’anticonvulsivante lamotrigina, con un dosaggio di 100-300 mg/die, è empiricamente considerato il trattamento di prima linea per la SUNCT [113,136-140]. Altri due farmaci anticonvulsivanti, considerati trattamenti di seconda linea per la SUNCT sono il gabapentin 800-2700 mg/die [116,133,141] e il topiramato 50-300 mg/die [135,142-144]. Alcuni pazienti hanno occasionalmente riportato un beneficio con l’uso di corticosteroidi [116,121,129,130, 143,145-148]. Anche la lidocaina, sia per endovena che per spray nasale, è stata usata per il trattamento di SUNCT [130,142,146]. Anche la carbamazepina 600-1200 mg/die è stata utilizzata, con esiti variabili, dopo che altri farmaci si erano dimostrati inefficaci [109]. La mancanza di una risposta assoluta alla carbamazepina nella SUNCT supporta la visione che la nevralgia trigeminale e la SUNCT siano due entità distinte [149]. Nella gestione dei pazienti con SUNCT sono stati impiegati anche approcci chirurgici. L’utilizzo di diversi anestetici locali, finalizzato all’interruzione della trasmissione degli impulsi dolorosi attraverso le fibre nervose, si è dimostrato generalmente inefficace nel sopprimere gli attacchi spontanei, sebbene in una minoranza di casi sia stato più difficile precipitare gli attacchi toccando le aree trigger anestetizzate [115,130-132]. Sono state eseguite diverse procedure chirurgiche dirette sul nervo trigemino ottenendo un miglioramento dei sintomi [131,147,150-153]. Comunque, data l’esistenza di outcomes negativi e di complicazioni post operatorie [120,131,146], le procedure chirurgiche dovrebbero essere considerate solo come ultima possibilità dopo che le opzioni farmacologiche sono state esaurite. In letteratura è riportato solo un caso di un paziente con SUNCT resistente ai farmaci che ha ricevuto la DBS con impianto di un elettrodo nell'ipotalamo inferiore posteriore [154], con sollievo dal dolore di lunga durata senza effetti collaterali. Rilevante il fatto che l’interruzione della stimolazione senza che il paziente ne fosse messo a conoscenza, era seguita da ricomparsa del dolore, che poi scompariva gradualmente quando l'apparecchio veniva riattivato. Questo riscontro sembra escludere un effetto placebo [154].
Bibliografia
1. Goadsby PJ, Lipton RB. A review of paroxysmal hemicranias, SUNCT syndrome and other short-lasting headaches with autonomic feature, including new cases. Brain 1997;120(Pt 1): 193-209.
2. The international classification of headache disorders, 2nd edition. Cephalalgia 2004;24(Suppl 1):9-160.
3. D'Alessandro R, Gamberini G, Benassi G, Morganti G, Cortelli P, Lugaresi E. Cluster headache in the Republic of San Marino. Cephalalgia 1986, 6:159-162.
4. Katsarava Z, Obermann M, Yoon MS, Dommes P, Kuznetsova J, Weimar C, Diener HC. Prevalence of cluster headache in a population-based sample in Germany. Cephalalgia 2007,
27:1014-1019. Epub 2007 Jul 1030
5. Sjaastad O, Bakketeig LS. Cluster headache prevalence. Vaga study of headache epidemiology. Cephalalgia 2003, 23:528-533.
6. Swanson JW, Yanagihara T, Stang PE, O'Fallon WM, Beard CM, Melton LJ 3rd, Guess HA. Incidence of cluster headaches: a population-based study in Olmsted County, Minnesota. Neurology 1994, 44:433-437.
7. Tonon C, Guttmann S, Volpini M, Naccarato S, Cortelli P, D'Alessandro R. Prevalence and incidence of cluster headache in the Republic of San Marino. Neurology 2002, 58:1407-1409.
8. Torelli P, Beghi E, Manzoni GC. Cluster headache prevalence in the Italian general population. Neurology 2005, 64:469-474.
9. Torelli P, Castellini P, Cucurachi L, Devetak M, Lambru G, Manzoni GC. Cluster headache prevalence: methodological considerations. A review of the literature. Acta Biomed 2006, 77:4-9.
10. Ekbom K, Ahlborg B, Schele R. Prevalence of migraine and cluster headache in Swedish men of 18. Headache 1978, 18:9-19.
11. Ekbom K, Svensson DA, Pedersen NL, Waldenlind E. Lifetime prevalence and concordance risk of cluster headache in the Swedish twin population. Neurology 2006, 67:798-803.
12. Evers S, Fischera M, May A, Berger K. Prevalence of cluster headache in Germany: results of the epidemiological DMKG study. J Neurol Neurosurg Psychiatry 2007, 78:1289-1290.
13. Lampl C. Childhood-onset cluster headache. Pediatr Neurol 2002, 27:138-140.
14. Garrido C, Tuna A, Ramos S, Temudo T. [Cluster headache in a 3 year old child]. Rev Neurol 2001, 33:732-735.
15. Manzoni GC. Male preponderance of cluster headache is progressively decreasing over the years. Headache 1997, 37:588-589.
16. Broner SW, Sun-Edelstein C, Lay CL. Cluster headache in women. Curr Pain Headache Rep 2007, 11:127-130.
17. Bahra A, May A, Goadsby PJ. Cluster headache: a prospective clinical study with diagnostic implications. Neurology 2002, 58:354-361.
18. Ekbom K. Evaluation of clinical criteria for cluster headache with special reference to the classification of the International Headache Society. Cephalalgia 1990, 10:195-197.
19. Cirillo M, Stellato D, Lombardi C, De Santo NG, Covelli V. Headache and cardiovascular risk factors: positive association with hypertension. Headache 1999, 39:409-416.
20. Kudrow L. The cyclic relationship of natural illumination to cluster period frequency. Cephalalgia 1987, 7(Suppl 6):76-78.
21. Donnet A, Lanteri-Minet M, Guegan-Massardier E, Mick G, Fabre N, Geraud G, Lucas C, Navez M, Valade D. Chronic cluster headache: a French clinical descriptive study. J Neurol Neurosurg Psychiatry 2007, 78:1354-1358. Epub 2007 Apr 1318
22. Torelli P, Manzoni GC. Pain and behaviour in cluster headache. A prospective study and review of the literature. Funct Neurol 2003, 18:205-210.
23. Ekbom K. A clinical comparison of cluster headache and migraine. Acta Neurol Scand 1970:1+.
24. Russell MB. Cluster headache sine headache: two new cases in one family. Cephalalgia 2002, 22:1.
25. Salvesen R. Cluster headache sine headache: case report. Neurology 2000, 55:451.
26. Boes C. Differentiating paroxysmal hemicrania from cluster headache. Cephalalgia 2005, 25:241-243.
27. Goadsby P, Lipton R. A review of paroxysmal hemicranias, SUNCT syndrome, and other short lasting headaches with autonomic features, including new cases. Brain 1997:193-209.
28. Goadsby PJ. Trigeminal autonomic cephalalgias. Pathophysiology and classification. Rev Neurol (Paris) 2005, 161:692-695.
29. Rapoport AM, Bigal ME. Hemicrania continua: clinical and nosographic update. Neurol Sci 2003, 24(Suppl 2):S118-121.
30. Silberstein SD, Peres MF. Hemicrania continua. Arch Neurol 2002, 59:1029-1030.
31. Treatment of acute cluster headache with sumatriptan. The Sumatriptan Cluster Headache Study Group. N Engl J Med 1991:322-326.
32. Schuh-Hofer S, Reuter U, Kinze S, Einhaupl K, Arnold G. Treatment of acute cluster headache with 20 mg sumatriptan nasal spray – an open pilot study. J Neurol 2002:94-99.
33. van Vliet JA, Bahra A, Martin V, Ramadan N, Aurora SK, Mathew NT, Ferrari MD, Goadsby PJ. Intranasal sumatriptan in cluster headache: randomized placebo-controlled double-blind study. Neurology 2003, 60:630-633.
34. Rapoport AM, Mathew NT, Silberstein SD, Dodick D, Tepper SJ, Sheftell FD, Bigal ME. Zolmitriptan nasal spray in the acute treatment of cluster headache: a double-blind study. Neurology 2007, 69:821-826.
35. Centonze V, Bassi A, Causarano V, Dalfino L, Cassiano MA, Centonze A, Fabbri L, Albano O. Sumatriptan overuse in episodic cluster headache: lack of adverse events, rebound syndromes, drug dependence and tachyphylaxis. Funct Neurol 2000, 15:167-170.
36. Paemeleire K, Evers S, Goadsby PJ. Medication-overuse headache in patients with cluster headache. Curr Pain Headache Rep 2008, 12:122-127.
37. Fogan L. Treatment of cluster headache. A double-blind comparison of oxygen v air inhalation. Arch Neurol 1985:362-363.
38. Kudrow L. Response of cluster headache attacks to oxygen inhalation. Headache 1981:1-4.
39. Ekbom K, Paalzow L, Waldenlind E. Low biological availability of ergotamine tartrate after oral dosing in cluster headache. Cephalalgia 1981, 1:203-207.
40. Bussone G, Leone M, Peccarisi C, Micieli G, Granella F, Magri M, Manzoni GC, Nappi G. Double blind comparison of lithium and verapamil in cluster headache prophylaxis. Headache 1990, 30:411-417.
41. Leone M, D'Amico D, Frediani F, Moschiano F, Grazzi L, Attanasio A, Bussone G. Verapamil in the prophylaxis of episodic cluster headache: a double-blind study versus placebo. Neurology 2000:1382-1385.
42. Cohen AS, Matharu MS, Goadsby PJ. Electrocardiographic abnormalities in patients with cluster headache on verapamil therapy. Neurology 2007, 69:668-675.
43. Leone M, Dodick D, Rigamonti A, D'Amico D, Grazzi L, Mea E, Bussone G. Topiramate in cluster headache prophylaxis: an open trial. Cephalalgia 2003, 23:1001-1002.
44. McGeeney BE. Topiramate in the treatment of cluster headache. Curr Pain Headache Rep 2003, 7:135-138.
45. Reilly D. Lithium vs placebo in cluster headache. Cephalalgia 1998, 18:1.
46. Steiner T, Hering R, Couturier E, Davies P, Whitmarsh T. Doubleblind placebo-controlled trial of lithium in episodic cluster headache. Cephalalgia 1997:673-675.
47. Pascual J, Lainez MJ, Dodick D, Hering-Hanit R. Antiepileptic drugs for the treatment of chronic and episodic cluster headache: a review. Headache 2007, 47:81-89.
48. Leandri M, Luzzani M, Cruccu G, Gottlieb A. Drug-resistant cluster headache responding to gabapentin: a pilot study. Cephalalgia 2001, 21:744-746.
49. Schuh-Hofer S, Israel H, Neeb L, Reuter U, Arnold G. The use of gabapentin in chronic cluster headache patients refractory to first-line therapy. Eur J Neurol 2007, 14:694-696.
50. Hering-Hanit R, Gadoth N. The use of baclofen in cluster headache. Curr Pain Headache Rep 2001, 5:79-82.
51. Leone M, D'Amico D, Moschiano F, Fraschini F, Bussone G. Melatonin versus placebo in the prophylaxis of cluster headache: a double-blind pilot study with parallel groups. Cephalalgia
1996, 16:494-496.
52. Pringsheim T, Magnoux E, Dobson CF, Hamel E, Aube M. Melatonin as adjunctive therapy in the prophylaxis of cluster headache: a pilot study. Headache 2002, 42:787-792.
53. Hassenbusch SJ, Kunkel RS, Kosmorsky GS, Covington EC, Pillay PK. Trigeminal cisternal injection of glycerol for treatment of chronic intractable cluster headaches. Neurosurgery 1991, 29:504-508.
54. Pieper DR, Dickerson J, Hassenbusch SJ. Percutaneous retrogasserian glycerol rhizolysis for treatment of chronic intractable cluster headaches: long-term results. Neurosurgery 2000,
46:363-368. discussion 368–370
55. Donnet A, Tamura M, Valade D, Regis J. Trigeminal nerve radiosurgical treatment in intractable chronic cluster headache: unexpected high toxicity. Neurosurgery 2006, 59:1252-1257.
56. Donnet A, Valade D, Regis J. Gamma knife treatment for refractory cluster headache: prospective open trial. J Neurol Neurosurg Psychiatry 2005, 76:218-221.
57. McClelland S 3rd, Barnett GH, Neyman G, Suh JH. Repeat trigeminal nerve radiosurgery for refractory cluster headache fails to provide long-term pain relief. Headache 2007, 47:298-300.
58. McClelland S 3rd, Tendulkar RD, Barnett GH, Neyman G, Suh JH. Long-term results of radiosurgery for refractory cluster headache. Neurosurgery 2006, 59:1258-1262. discussion 1262-1253
59. Leone M, Franzini A, Broggi G, Bussone G. Hypothalamic deep brain stimulation for intractable chronic cluster headache: a 3-year follow-up. Neurol Sci 2003, 24(Suppl 2):S143-145.
60. Leone M, Franzini A, Broggi G, Bussone G. Hypothalamic stimulation for intractable cluster headache: long-term experience. Neurology 2006, 67:150-152.
61. Schoenen J, Di Clemente L, Vandenheede M, Fumal A, De Pasqua V, Mouchamps M, Remacle JM, de Noordhout AM. Hypothalamic stimulation in chronic cluster headache: a pilot study of efficacy and mode of action. Brain 2005, 128:940-947. Epub 2005 Feb 2002
62. Bartsch T, Pinsker MO, Rasche D, Kinfe T, Hertel F, Diener HC, Tronnier V, Mehdorn HM, Volkmann J, Deuschl G, Krauss JK. Hypothalamic deep brain stimulation for cluster headache:
experience from a new multicase series. Cephalalgia 2008, 28:285-295.
63. Pinsker MO, Bartsch T, Falk D, Volkmann J, Herzog J, Steigerwald F, Diener HC, Deuschl G, Mehdorn M. Failure of deep brain stimulation of the posterior inferior hypothalamus in chronic cluster headache – report of two cases and review of the literature. Zentralbl Neurochir 2008, 69:76-79.
64. Burns B, Watkins L, Goadsby PJ. Treatment of medically intractable cluster headache by occipital nerve stimulation: longterm follow-up of eight patients. Lancet 2007, 369:1099-1106.
65. Leone M, Franzini A, Cecchini AP, Broggi G, Bussone G. Stimulation of occipital nerve for drug-resistant chronic cluster headache. Lancet Neurol 2007, 6:289-291.
66. Schwedt TJ, Dodick DW, Trentman TL, Zimmerman RS. Occipital nerve stimulation for chronic cluster headache and hemicrania continua: pain relief and persistence of autonomic features. Cephalalgia 2006, 26:1025-1027.
67. Antonaci F, Sjaastad O. Chronic paroxysmal hemicrania (CPH): a review of the clinical manifestations. Headache 1989;29: 648-56.
68. Lance JW, Goadsby P. Mechanism and management of headache. Philadelphia: Elsevier; 2005
69. Siow HC. Seasonal episodic paroxysmal hemicrania responding to cyclooxygenase-2 inhibitors. Cephalalgia 2004;24:414-5.
70. Rossi P, Di Lorenzo G, Faroni J, Sauli E. Seasonal, extratrigeminal, episodic paroxysmal hemicrania successfully treated with single suboccipital steroid injections. Eur J Neurol 2005;12:
903-6.
71. Boes CJ, Dodick DW. Refining the clinical spectrum of chronic paroxysmal hemicrania: a review of 74 patients. Headache 2002;42:699-708.
72. Sjaastad O. Chronic Paroxysmal Hemicrania (CPH). In: Vinken PJ, Bruyn GK, Klawans HL, Rose FC, editors. Handbook of clinical neurology. Amsterdam: Elsevier Science; 1986. p. 257-66.
73. Newman LC, Goadsby P. The paroxysmal hemicranias, SUNCT syndrome, and hypnic headache. In: Silberstein SD, Lipton RB, Dalessio DJ, editors. Wolff’s headache and other head pain, Oxford: Oxford University Press; 2001. p. 310-24.
74. Talvik I, Koch K, Kolk A, Talvik T. Chronic paroxysmal hemicrania in a 3-year, 10-month-old female. Pediatr Neurol 2006;34:225-7.
75. de Almeida DB, Cunali PA, Santos HL, Brioschi M, Prandini M. Chronic paroxysmal hemicrania in early childhood: case report. Cephalalgia 2004;24:608-9.
76. Broeske D, Lenn NJ, Cantos E. Chronic paroxysmal hemicrania in a young child: possible relation to ipsilateral occipital infarction. J Child Neurol 1993;8:235-6.
77. Cohen AS, Matharu MS, Goadsby PJ. Paroxysmal hemicrania in a family. Cephalalgia 2006;26:486-8.
78. Pelz M, Merskey H. A case of pre-chronic paroxysmal hemicrania. Cephalalgia 1982;2:47-50.
79. Pareja JA. Chronic paroxysmal hemicrania: dissociation of the pain and autonomic features. Headache 1995;35:111-3.
80. Pollmann W, Pfaffenrath V. Chronic paroxysmal hemicrania: the first possible bilateral case. Cephalalgia 1986;6:55-7.
81. Newman LC, Lipton RB. Paroxysmal hemicranias. In: Goadsby P, Silberstein SD, editors. Headache. Boston: Butterworth-Heinemann; 1997. p. 243-50.
82. Boes CJ, Swanson JW. Paroxysmal hemicrania, SUNCT, and hemicrania continua. Semin Neurol 2006;26:260-70.
83. Castellanos-Pinedo F, Zurdo M, Martinez-Acebes E. Hemicrania continua evolving from episodic paroxysmal hemicrania. Cephalalgia 2006;26:1143-5.
84. Russell D. Chronic paroxysmal hemicrania: severity, duration and time of occurrence of attacks. Cephalalgia 1984;4:53-6.
85. Kayed K, Godtlibsen OB, Sjaastad O. Chronic paroxysmal hemicrania IV: “REM sleep locked” nocturnal headache attacks. Sleep 1978;1:91-5.
86. Bogucki A, Szymanska R, Braciak W. Chronic paroxysmal hemicrania: lack of pre-chronic stage. Cephalalgia 1984;4:187-9.
87. Sjaastad O, Russell D, Saunte C, Horven I. Chronic paroxysmal hemicrania. VI. Precipitation of attacks. Further studies on the precipitation mechanism. Cephalalgia 1982;2:211-4.
88. Sjaastad O, Saunte C, Graham JR. Chronic paroxysmal hemicrania. VII. Mechanical precipitation of attacks: new cases and localization of trigger points. Cephalalgia 1984;4:113-8.
89. Matharu MS, Boes CJ, Goadsby PJ. Management of trigeminal autonomic cephalgias and hemicrania continua. Drugs 2003;63: 1637-77.
90. Antonaci F, Pareja JA, Caminero AB, Sjaastad O. Chronic paroxysmal hemicrania and hemicrania continua. Parenteral indomethacin: the “indotest.” Headache 1998;38:122-8.
91. Warner JS, Wamil AW, McLean MJ. Acetazolamide for the treatment of chronic paroxysmal hemicrania. Headache 1994;34:597-9.
92. Pareja J, Sjaastad O. Chronic paroxysmal hemicrania and hemicrania continua. Interval between indomethacin administration and response. Headache 1996;36:20-3.
93. Sjaastad O, Stovner LJ, Stolt-Nielsen A, Antonaci F, Fredriksen TA. CPH and hemicrania continua: requirements of high indomethacin dosages—an ominous sign? Headache 1995;35:363-7.
94. Sjaastad O, Apfelbaum R, Caskey W, Christoffersen B, Diamond S, Graham J, et al. Chronic paroxysmal hemicrania (CPH). The clinical manifestations. A review. Ups J Med Sci Suppl 1980;31:27-33.
95. Pareja JA, Caminero AB, Franco E, Casado JL, Pascual J, Sanchez del Rio M. Dose, efficacy and tolerability of long-term indomethacin treatment of chronic paroxysmal hemicrania and
hemicrania continua. Cephalalgia 2001;21:906-10.
96. Sjaastad O, Antonaci F. Chronic paroxysmal hemicrania: a case report. Long-lasting remission in the chronic stage. Cephalalgia 1987;7:203-5.
97. Benoliel R, Sharav Y. Paroxysmal hemicrania. Case studies and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998;85:285-92.
98. Hannerz J, Ericson K, Bergstrand G. Chronic paroxysmal hemicrania: orbital phlebography and steroid treatment. A case report. Cephalalgia 1987;7:189-92.
99. Evers S, Husstedt IW. Alternatives in drug treatment of chronic paroxysmal hemicrania. Headache 1996;36:429-32.
100. Mathew NT, Kailasam J, Fischer A. Responsiveness to celecoxib in chronic paroxysmal hemicrania. Neurology 2000;55:316.
101. Coria F, Claveria LE, Jimenez-Jimenez FJ, de Seijas EV. Episodic paroxysmal hemicrania responsive to calcium channel blockers. J Neurol Neurosurg Psychiatry 1992;55:166.
102. Shabbir N, McAbee G. Adolescent chronic paroxysmal hemicrania responsive to verapamil monotherapy. Headache 1994;34: 209-10.
103. Pascual J, Quijano J. A case of chronic paroxysmal hemicrania responding to subcutaneous sumatriptan. J Neurol Neurosurg Psychiatry 1998;65:407.
104. Dahlof C. Subcutaneous sumatriptan does not abort attacks of chronic paroxysmal hemicrania (CPH). Headache 1993;33:201-2.
105. Antonaci F, Pareja JA, Caminero AB, Sjaastad O. Chronic paroxysmal hemicrania and hemicrania continua: lack of efficacy of sumatriptan. Headache 1998;38:197-200.
106. Cohen AS, Goadsby PJ. Paroxysmal hemicrania responding to topiramate. J Neurol Neurosurg Psychiatry 2007;78:96-7.
107. Pareja JA, Cuadrado ML. SUNCT syndrome: an update. Expert Opin Pharmacother 2005;6: 591-9.
108. Pareja JA, Sjaastad O. SUNCT syndrome. A clinical review. Headache 1997;37:195-202.
109. Matharu MS, Cohen AS, Boes CJ, Goadsby PJ. Short-lasting unilateral neuralgiform headache with conjunctival injection and tearing syndrome: a review. Curr Pain Headache Rep 2003;7:
308-18.
110. Sekhara T, Pelc K, Mewasingh LD, Boucquey D, Dan B. Pediatric SUNCT syndrome. Pediatr Neurol 2005;33:206-7.
111. Vikelis M, Xifaras M, Mitsikostas DD. SUNCT syndrome in the elderly. Cephalalgia 2005;25:1091-2.
112. Gantenbein AR, Goadsby PJ. Familial SUNCT. Cephalalgia 2005;25:457-9.
113. D Andrea G, Granella F, Ghiotto N, Nappi G. Lamotrigine in the treatment of SUNCT syndrome. Neurology 2001;57:1723-5.
114. D Andrea G, Granella F. SUNCT syndrome: the first case in childhood. Short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing. Cephalalgia 2001; 21:701-2.
115. Sabatowski R, Huber M, Meuser T, Radbruch L. SUNCT syndrome: a treatment option with local opioid blockade of the superior cervical ganglion? A case report. Cephalalgia 2001; 21:154-6.
116. Graff-Radford SB. SUNCT syndrome responsive to gabapentin (Neurontin). Cephalalgia 2000;20:515-7.
117. Wingerchuk DM, Nyquist PA, Rodriguez M, Dodick DW. Extratrigeminal, short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT): new pathophysiologic entity or variation on a theme? Cephalalgia 2000; 20:127-9.
118. Cohen AS, Matharu MS, Goadsby PJ. Short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing (SUNCT) or cranial autonomic features (SUNA)–a prospective clinical study of SUNCT and SUNA. Brain 2006; 129:2746-60.
119. Jimenez-Huete A, Franch O, Pareja JA. SUNCT syndrome: priming of symptomatic periods and worsening of symptoms by treatment with calcium channel blockers. Cephalalgia 2002; 22:812-4.
120. Black DF, Dodick DW. Two cases of medically and surgically intractable SUNCT: a reason for caution and an argument for a central mechanism. Cephalalgia 2002;22:201-4.
121. Montes E, Alberca R, Lozano P, Franco E, Martinez-Fernandez E, Mir P. Statuslike SUNCT in two young women. Headache 2001;41:826-9.
122. Pareja JA, Joubert J, Sjaastad O. SUNCT syndrome. Atypical temporal patterns. Headache 1996;36:108-110.
123. Pareja JA, Shen JM, Kruszewski P, Caballero V, Pamo M, Sjaastad O. SUNCT syndrome: duration, frequency, and temporal distribution of attacks. Headache 1996;36:161-5.
124. Kruszewski P, Fasano ML, Brubakk AO, Shen JM, Sand T, Sjaastad O. Shortlasting, unilateral, neuralgiform headache attacks with conjunctival injection, tearing, and subclinical forehead sweating (“Sunct” syndrome): II. Changes in heart rate and arterial blood pressure during pain paroxysms. Headache 1991; 31:399-405.
125. Kruszewski P, White LR, Shen JM, Pareja JA, Zhao JM, Schaanning J, et al. Respiratory studies in SUNCT syndrome. Headache 1995;35:344-8.
126. Sjaastad O, Saunte C, Salvesen R, Fredriksen TA, Seim A, Roe OD, et al. Shortlasting unilateral neuralgiform headache attacks with conjunctival injection, tearing, sweating, and rhinorrhea. Cephalalgia 1989;9:147-56.
127. Bouhassira D, Attal N, Esteve M, Chauvin M. “SUNCT” syndrome. A case of transformation from trigeminal neuralgia? Cephalalgia 1994;14:168-70.
128. Benoliel R, Sharav Y. Trigeminal neuralgia with lacrimation or SUNCT syndrome? Cephalalgia 1998;18:85-90.
129. Calvo JF, Bruera OC, de Lourdes Figuerola M, Gestro D, Tinetti N, Leston JA. SUNCT syndrome: clinical and 12-year follow-up case report. Cephalalgia 2004;24:900-2.
130. Pareja JA, Kruszewski P, Sjaastad O. SUNCT syndrome: trials of drugs and anesthetic blockades. Headache 1995;35:138-42.
131. Hannerz J, Linderoth B. Neurosurgical treatment of short-lasting, unilateral, neuralgiform hemicrania with conjunctival injection and tearing. Br J Neurosurg 2002;16:55-8.
132. De Benedittis G. SUNCT syndrome associated with cavernous angioma of the brain stem. Cephalalgia 1996;16:503-6.
133. Hunt CH, Dodick DW, Bosch EP. SUNCT responsive to gabapentin. Headache 2002;42:525-6.
134. Hannerz J, Greitz D, Hansson P, Ericson K. SUNCT may be another manifestation of orbital venous vasculitis. Headache 1992;32:384-9.
135. Sesso RM. SUNCT syndrome or trigeminal neuralgia with lacrimation and conjunctival injection? Cephalalgia 2001;21:151-3.
136. D Andrea G, Granella F, Cadaldini M. Possible usefulness of lamotrigine in the treatment of SUNCT syndrome. Neurology 1999;53:1609.
137. Leone M, Rigamonti A, Usai S, Damico D, Grazzi L, Bussone G. Two new SUNCT cases responsive to lamotrigine. Cephalalgia 2000;20:845-7.
138. Gutierrez-Garcia JM. SUNCT syndrome responsive to lamotrigine. Headache 2002;42:823-5.
139. Malik K, Rizvi S, Vaillancourt PD. The SUNCT syndrome: successfully treated with lamotrigine. Pain Med 2002;3:167-8.
140. Chakravarty A, Mukherjee A. SUNCT syndrome responsive to lamotrigine: documentation of the first Indian case. Cephalalgia 2003;23:474-5.
141. Porta-Etessam J, Benito-Leon J, Martinez-Salio A, Berbel A. Gabapentin in the treatment of SUNCT syndrome. Headache 2002;42:523-4.
142. Matharu MS, Boes CJ, Goadsby PJ. SUNCT syndrome: prolonged attacks, refractoriness and response to topiramate. Neurology 2002;58:1307.
143. Rossi P, Cesarino F, Faroni J, Malpezzi MG, Sandrini G, Nappi G. SUNCT syndrome successfully treated with topiramate: case reports. Cephalalgia 2003;23:998-1000.
144. Kuhn J, Vosskaemper M, Bewermeyer H. SUNCT syndrome: a possible bilateral case responding to topiramate. Neurology 2005; 64:2159.
145. Pareja JA, Pareja J, Palomo T, Caballero V, Pamo M. SUNCT syndrome: repetitive and overlapping attacks. Headache 1994; 34:114-6.
146. Matharu MS, Cohen AS, Goadsby PJ. SUNCT syndrome responsive to intravenous lidocaine. Cephalalgia 2004;24:985-92.
147. Morales-Asin F, Espada F, Lopez-Obarrio LA, Navas I, Escalza I, Iniguez C. A SUNCT case with response to surgical treatment. Cephalalgia 2000;20:67-8.
148. Pareja JA, Cuadrado ML, Caminero AB, Barriga FJ, Baron M, Sanchez-del-Rio M. Duration of attacks of first division trigeminal neuralgia. Cephalalgia 2005;25:305-8.
149. Sjaastad O, Kruszewski P. Trigeminal neuralgia and “SUNCT” syndrome: similarities and differences in the clinical pictures. An overview. Funct Neurol 1992;7:103-7.
150. Sprenger T, Valet M, Platzer S, Pfaffenrath V, Steude U, Tolle TR. SUNCT: bilateral hypothalamic activation during headache attacks and resolving of symptoms after trigeminal decompression. Pain 2005;113:422-6.
151. Gardella L, Viruega A, Rojas H, Nagel J. A case of a patient with SUNCT syndrome treated with Jannetta procedure. Cephalalgia 2001;21:996-9.
152. Lenarest M, Diederich N, Phuoe D. A patient with SUNCT cured by the Jannetta procedure [letter]. Cephalalgia 1997;17:460.
153. Ertsey C, Bozsik G, Afra J. A case of SUNCT syndrome with neurovascular compression [abstract]. Cephalalgia 2000;20:325.
154. Leone M, Franzini A, D Andrea G, Broggi G, Casucci G, Bussone G. Deep brain stimulation to relieve drug-resistant SUNCT. Ann Neurol 2005;57:924-7.